|
CF is caused by the abnormal functioning of the protein CF transmembrane conductance regulator (CFTR). CFTR is only expressed in a number of cells but all cells carry the gene for the protein. CFTR is mainly found in epithelial cells and in order for CFTR to be expressed a certain amount must be produced at the right time. The CFTR protein contains 1480 amino acids and is important in regulating the flow of chloride and sodium across the epithelial surface. The function of CFTR is disrupted by changes in the base sequence, these changes are called mutations. Over 1,200 mutations have been discovered for CF but the most common is deltaF508-which is a 3 base pair deletion (of amino acid phenylalanine) and account for 76% of affected chromosomes in the UK. CF is an autosomal recessive disease, so both CFTR genes are required for the individual to be diagnosed with CF, if only one is present then the individual will be a carrier. A classification system has been produced that characterises CFTR mutation depending on the effects the mutation has on the CFTR production. Class 1 are nonsense mutations as there is a premature stop codon which leads to the production of unstable mRNA or the release of incomplete proteins that are not functional. This protein is degraded before it can reach the membrane. The phenotype of patients carrying the stop mutation is severe. In Class 2 after CFTR has been translated into a peptide in the ribosome, it will go through a series of processes in the ER and Golgi apparatus. These processes are glycolisation and folding by chaperons that enable the trafficking of CFTR to the apical cell membrane. In Class 2 mutations, the CFTR protein is unable to fold correctly due to the deltaF508. The protein is retained in the ER and eventually targeted for degradation. Class 3- phosphorylation of CFTR protein by protein kinase (PK) & dephosphorylation by protein phosphatase (PP) is important in regulating CFTR chloride channel activity. Phosphorylation of the regulatory domain causes ATP to bind to the nucleotide binding domain (NBD) resulting in induction of chloride transport. In Class 3 CFTR is produced, processed and transported and inserted into the apical membrane however phosphorylation or binding of ATP does not occur. Class 4- in this class of mutation the CFTR protein is produced, processed, transported to the apical membrane. Phosphorylation and dephosphorylation also occur. In class 4, phosphorylation actually reduces chloride transport. Class 5- this class of mutations generates both abnormal and correctly spliced transcripts. These patients have a mild phenotype but with variable disease expression depending on the level of correctly spliced transcripts. Steps of diagnosing a baby with CF Neonatal screening test To diagnose a baby with CF the first test that is to be performed is the neonatal screening test. During the 1st 2 weeks of Childs life, high levels of IRT levels will be shown if CF is present. In this test a blood sample is taken from the child heel and the level of this pro-enzyme is monitored. If levels are high, then other genetic test can be carried out to find out exactly which mutations are present and detect the severity of the disease. This test is not reliable after 2 weeks of birth as several other factors may play a part in the high levels of IRT. PCR Analysis One of the 1st genetic tests that can be carried out for the diagnosis of CF is PCR analysis. This test is carried out to see if the most common mutation (deltaF508) is present which is the deletion of 3bp. PCR primers have been developed that can distinguish a normal gene from a mutant gene. With these primers a 154 bp product is produced from a normal individual and a 151 bp product is amplified from DNA of an individual with the disease. If this CF mutation is present it will show a distinct pattern from a normal pattern. If both genes have this mutation, two bands will be seen. If 1 band is present, this indicates that other the second gene has a different mutation which can be followed up by other genetic tests. Normal Carrier CF Individual Individual Individual ---------------------------------- 154 bp ___2x ___ 151 bp ___ ___2x ---------------------------------- ARMS Another test that can be performed is the amplification refractory mutation system (ARMS). This can be used to recognize the 20 most common CF mutations. In this technique, three multiplex PCR reactions are performed in parallel for each individual. Each tube contains number of separate primers capable of detecting different mutations. In each tube, sets of primer pairs are designed to anneal a particular allele to produce distinct band. A band is only produced in presence of mutant allele and the absence of the band indicates that the allele is absent; therefore positive signals are easily differentiated in order to identify the exact mutations present. Since it is possible for one of the tubes to produce no bands, there are always positive controls to show that the PCR reaction is functioning properly in each tube. The ARMS assay is accurate, rapid and easy to perform, but it does not distinguish between homozygotes and heterozygotes except for the DF508 mutation. Multiplex Allele Specific PCR Allele specific oligonucleotide (ASO) dot-blot is a widely used technique to diagnose CF. Genomic DNA from the patient is amplified by PCR and transferred onto nylon membranes as a dot-blot. Membranes are hybridized with either a radiolabelled wild type allele specific oligonucleotide (ASO) or mutated ASO. Following autoradiographic exposure, the combination of oligonucleotide hybridization is observed. Using this combination an ASO dot-blot can clearly distinguish homozygous, heterozygous and wild type subjects. 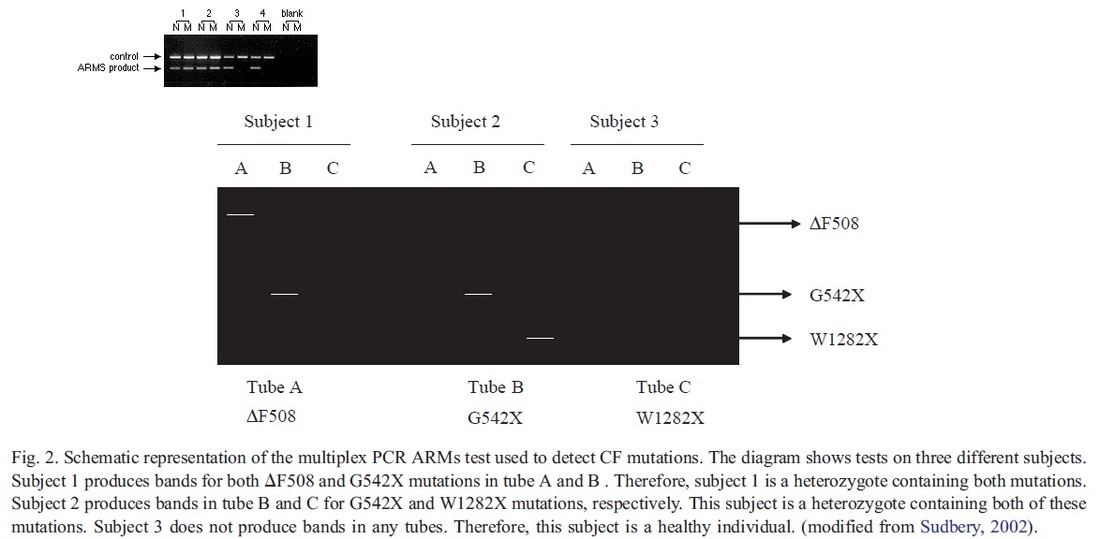 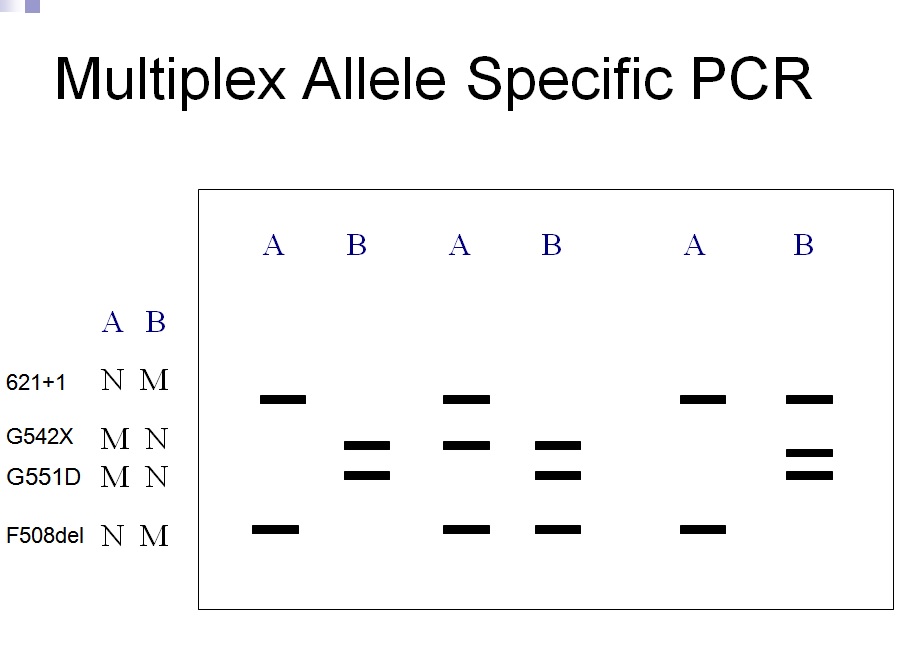
1 Comment
|
A GOOD HEALTH MAKES YOU RICHHealth is crucial in every single person’s life. Its something that money can’t buy. Thus a good health makes you rich so look after it. Archives
May 2017
Categories |
Welcome
 RSS Feed
RSS Feed
© COPYRIGHT 2024. ALL RIGHTS RESERVED.
Proudly powered by Weebly